Sickle cell anemia
Tanja Unterberger studied journalism and communication science in Vienna. In 2015 she started her work as a medical editor at in Austria. In addition to writing specialist texts, magazine articles and news, the journalist also has experience in podcasting and video production.
More about the experts All content is checked by medical journalists.Sickle cell disease (also sickle cell disease or Drepanocytosis) is a hereditary disease in which the red blood cells become sickle-shaped. The cause is a genetic defect. Symptoms include circulatory disorders and anemia. A cure is currently only possible with a stem cell transplant. Read more about causes, symptoms and therapy here!
ICD codes for this disease: ICD codes are internationally recognized codes for medical diagnoses. They can be found, for example, in doctor's letters or on certificates of incapacity for work. D57

Brief overview
- Description: Hereditary disease in which the red blood cells (erythrocytes) become sickle-shaped
- Causes: The cause of sickle cell anemia is a defective gene that is responsible for the formation of hemoglobin (red blood pigment).
- Prognosis: Sickle cell anemia has a varying degree of severity. The sooner the symptoms are treated, the better the prognosis. If left untreated, the disease is usually fatal.
- Symptoms: Severe pain, circulatory disorders, anemia, frequent infections, organ damage (e.g. spleen), strokes, growth disorders
- Treatment: The symptoms of sickle cell disease can be relieved with medication (e.g. hydroxycarbamide, pain relievers) and blood transfusions. A cure is possible with a stem cell transplant.
- Diagnosis: conversation with the doctor, physical examination, blood test, ultrasound, CT, MRI
What is sickle cell anemia?
Sickle cell disease - also known as sickle cell disease (SCD) or drepanocytosis - is a hereditary disease. During the disease, healthy red blood cells (erythrocytes) change into abnormal, sickle-shaped cells (sickle cells). Because of their shape, these can clog blood vessels in the body. Typical symptoms of the disease are severe pain, circulatory disorders, anemia and organ damage.
Sickle cell anemia is caused by an altered gene that is responsible for the formation of the red blood pigment (hemoglobin). Sickle cell disease is not contagious and already exists at birth. The first symptoms often appear in infancy or toddlerhood.
The disease belongs to the group of hemoglobinopathies. These are various disorders of the red blood pigment hemoglobin.
Doctors prefer the term sickle cell disease instead of sickle cell anemia, as not all forms are associated with anemia. In addition, the anemia is not the main symptom of the disease, but the symptoms that trigger the vascular occlusion.
How common is sickle cell anemia?
Sickle cell anemia is one of the most common hereditary diseases and the most common blood disease in the world. There are currently around 3,000 to 5,000 people in Germany with sickle cell disease. According to an estimate by the World Health Organization (WHO), over 300,000 children are born with sickle cell disease worldwide every year.
Who is particularly affected?
Sickle cell anemia mainly affects people from Central and West Africa. It initially occurred mainly in the sub-Saharan part of the African continent. However, due to migration, sickle cell disease has meanwhile become widespread worldwide. Today, many people from parts of the Mediterranean, the Middle East, India and North America are also affected.
Sickle cell disease has also been widespread in Northern Europe (e.g. Germany, Austria, France, England, the Netherlands, Belgium and Scandinavia) since the 1960s.
Sickle cell anemia and malaria
People with sickle cell disease are less prone to malaria. The reason for this: The red blood cells normally transport the malaria pathogens (plasmodia) through the body via the blood. In sickle cell disease, however, the red blood cells change to sickle cells and are less mobile. The malaria pathogens therefore survive worse.
This gives people who have a healthy and a diseased gene (so-called heterozygous gene carriers) a survival advantage (heterozygous advantage) in the regions in which malaria spreads. This is also the explanation for why a relatively large number of people there are sick with sickle cell anemia.
How does sickle cell anemia develop?
Sickle cell anemia is a hereditary disease. The cause is a changed gene (mutation) that the father and mother transmit to their child. The genetic defect changes healthy, red blood pigment (hemoglobin; Hb for short) into sickle-cell hemoglobin (hemoglobin S; HbS for short).
Congenital genetic defect
The red blood cells have the vital task of transporting oxygen from the lungs to all parts of the body. To do this, the oxygen binds to the red blood pigment. This is a protein found in red blood cells.
The red blood cells normally consist of “healthy” hemoglobin, which is made up of two protein chains - the alpha and beta chains. These chains make the blood cells round and smooth, which allows them to fit through every tiny blood vessel and provide all organs with vital oxygen and nutrients.
In sickle cell anemia, genetic errors (mutations) mean that the so-called beta chain of hemoglobin is abnormally changed (hemoglobin S).If there is too little oxygen in the blood, the shape of the sickle cell hemoglobin changes and with it that of the red blood cells.
The hemoglobin particles form rigid shapes and clump together, causing the red blood cells to take on the typical shape and look like small sickles. This is where the term "sickle cell disease" comes from.
Because of their shape, sickle cells are much more immobile and die earlier than healthy red blood cells (hemolysis). On the one hand, this results in anemia in the body (so-called corpuscular hemolytic anemia) because the person concerned has too few red blood cells. On the other hand, because of their shape, the sickle cells more easily clog smaller blood vessels.
This means that the affected areas of the body are no longer adequately supplied with oxygen. As a result, the tissue can die and, in the worst case, the organs can no longer fulfill their vital functions.
How is sickle cell disease inherited?
Sickle cell anemia is inherited from both father and mother. It is a so-called autosomal recessive inheritance: Each parent carries at least one pathological gene in the chromosomes (carrier of the genetic information).
It has different effects whether the child inherits only one pathological gene or both genes:
Homozygous gene carrier
If both the father and the mother carry the modified gene in them and they both pass it on to their child, the child carries two modified genes (homozygous gene carrier). It suffers from sickle cell anemia. In this case, the child only produces pathological hemoglobin (HbS) and no healthy hemoglobin (Hb).
Heterozygous gene carrier
If one parent passes on a healthy gene and the other parent passes on a diseased gene, the child will produce both healthy hemoglobin and sickle cell hemoglobin. In this case, the child will not get sickle cell anemia. However, it carries the pathological gene and is able to pass it on to its children later (heterozygous gene carrier).
Doctors also advise carriers of the disease to seek advice on the possible inheritance and the associated risks for the child if they wish to have children
Is sickle cell disease curable?
The prognosis of sickle cell anemia depends heavily on how well and early on the symptoms and complications are treated. Around 85 to 95 percent of all children with sickle cell disease reach adulthood in countries with good health care (e.g. Europe, USA). The mean life expectancy is then around 40 to 50 years. In countries with poorer medical care, mortality is higher.
With blood stem cell transplants, it is now possible for doctors to completely cure people with sickle cell anemia. However, this treatment also carries some risks and is therefore reserved for severe cases of sickle cell anemia.
How is the disease going?
Some people with sickle cell disease have few symptoms, while others suffer greatly from the effects of the disease. It has not yet been clarified why the disease progresses so differently.
People with sickle cell anemia usually show the first signs of the disease as a baby.
What types of sickle cell disease are there?
There are different forms of sickle cell anemia. These arise depending on how the gene that is responsible for the formation of the red blood pigment is changed. All forms are inherited. However, the respective forms run differently, and the symptoms are also differently pronounced.
The most common forms of sickle cell disease are:
Sickle Cell Disease HbSS (SCD-S / S)
SCD-S / S is the most common type of sickle cell anemia. It's the hardest. Those affected have each inherited a modified gene from both parents. Therefore, they only form the pathologically altered sickle cell hemoglobin (HbS).
Sickle cell disease HbSß-Thal (SCS-S / beta-Thal)
With SCS-S / beta-Thal, children inherit the sickle cell gene from one parent and a gene for what is known as beta thalassemia from the other. The latter is another blood disorder in which the body makes too little or no hemoglobin. This form of sickle cell disease is rarer and usually milder.
Sickle Cell Disease HbSC (SCD-S / C)
In SCD-S / C, one parent inherits the gene for sickle cell disease and the other parent inherits an altered hemoglobin gene, the HbC gene. High blood levels of the red blood pigment hemoglobin are typical. Since the blood is thicker as a result, complications such as circulatory disorders may arise. This form of sickle cell anemia is usually less severe, but in some cases it can lead to blindness or deafness.
Sickle cell disease develops not only when a child inherits two sickle cell genes, but also when the parents pass on a sickle cell gene combined with another pathologically altered hemoglobin gene (such as the HbC or thalassemia gene).
What are the symptoms?
The symptoms that occur with sickle cell anemia usually affect the entire body. In principle, it is possible that the sickle cells clog every blood vessel in the body. The most common symptoms of sickle cell disease are:
Strong pain
People with sickle cell disease usually have severe pain (so-called pain crises). Your body makes substances that cause pain when the sickle cells clog the vessels and thereby block the blood supply to the organs (especially the bone marrow) and tissues. The main pain in your stomach, bones, and joints is during a crisis.
Weather changes, lack of fluids, infections with fever and exhaustion often trigger this pain. Elderly people with sickle cell anemia are also more often affected by severe pain crises than children.
Some people with sickle cell disease have short-term pain, while others often spend long periods of time in the hospital and need pain medication.
Anemia
Sickle cells are less stable than healthy red blood cells and therefore die earlier (hemolysis). Sickle cells survive for about ten to 20 days, while red blood cells are usually broken down after about 120 days. If there are too few red blood cells in the body, anemia will develop over time and the concentration of the red blood pigment will decrease.
Organ damage
Because the blood vessels are blocked by the sickle cells, the affected areas of the body are no longer adequately supplied with blood (so-called sickle cell crises). As a result, the tissue does not receive enough oxygen and nutrients, which leads to its death over time. The affected organs are then no longer able to function properly.
This mostly affects the bone marrow, lungs, brain, spleen and gastrointestinal tract.
Infections
If sickle cell anemia affects the spleen in the first few years of life, patients are at high risk of serious infections. The reason for this is that the spleen plays an important role in fighting pathogenic germs (e.g. bacteria such as pneumococci and meningococci, which trigger pneumonia or meningitis) in the body.
Infections with fever are an emergency in people with sickle cell disease! Therefore, immediately consult a doctor if the body temperature is over 38.5 degrees Celsius!
Hand-foot syndrome
Because of the vascular occlusions, small children often experience pain, redness and swelling of the hands and feet (hand-foot syndrome). This is often the first sign that the child has sickle cell disease.
Stunted growth
Small children often experience pain, redness and swelling of the hands and feet because of the vascular occlusion (hand-foot syndrome). This is often the first sign that the child has sickle cell disease.
Bone necrosis
Because the sickle cells clog the blood vessels, tissue in joints or bones also dies over time (avascular bone necrosis). The hip joints and shoulder joints are often affected.
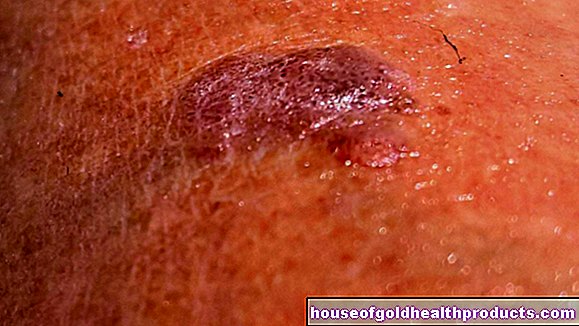
Ulcers
It is also possible that sickle cells clog the blood vessels in the skin (especially on the legs), which means that the tissue is no longer adequately supplied with nutrients. As a result, those affected often have painful, open wounds on their legs (ulcers) that are usually difficult to heal.
Visual disturbances and blindness
When the sickle cells clog blood vessels in the retina of the eyes, the surrounding tissue dies and scars form in the back of the eye. This affects the eyesight of those affected and, in the worst case, can lead to blindness.
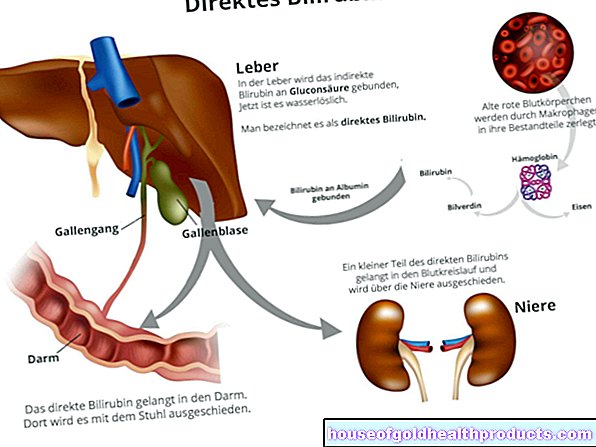
Gallstones, jaundice
Because the sickle-shaped red blood cells disintegrate more quickly in sickle cell anemia, more breakdown products are created in the blood, for example bilirubin (bile pigment). Bilirubin is produced when the red blood pigment hemoglobin is broken down. Sometimes these breakdown products form gallstones in the gallbladder. If these get stuck in the bile ducts, severe pain (biliary colic) and jaundice (jaundice) may result. Gallbladder inflammation is also possible as a complication of sickle cell disease.
Hemolytic crisis, aplastic crisis
In sickle cell disease, the sickle-shaped red blood cells are broken down more quickly. There is a risk of hemolytic crises, for example caused by infections, in which massive amounts of red blood cells break down. When no red blood cells are produced at all, doctors speak of an aplastic crisis. Those affected need immediate medical treatment! A blood transfusion is often necessary to prevent life-threatening oxygen starvation and cardiovascular failure.
Spleen Sequestration and Spleen Enlargement
The spleen is the organ in which the red blood cells are broken down. In people with sickle cell anemia, it is possible that large amounts of blood suddenly “sink” into the spleen (spleen sequestration). This sometimes leads to life-threatening anemia and a lack of oxygen.
Spleen sequestration usually develops over one to three days. Symptoms include fever and abdominal pain. Those affected are often pale and limp, similar to a cold. The spleen swells due to the amount of blood that sinks. In most cases this can be felt.
It is important that parents of babies and young children with sickle cell disease learn to palpate their child's spleen. If the spleen is enlarged, doctors advise parents to take their children to hospital immediately.
Acute Thoracic Syndrome (ATS)
When sickle cells clog blood vessels in the lungs, what is known as acute thoracic syndrome (ATS for short) can occur. In addition to spleen sequestration, acute thoracic syndrome is one of the most common causes of death in people with sickle cell disease. An acute chest syndrome usually arises as a result of a pain crisis.
The symptoms of acute chest syndrome are similar to those of pneumonia: those affected (often children) have a fever, cough, difficulty breathing, and severe chest pain, especially when breathing. If the thoracic syndrome occurs again and again, this damages the lungs in the long term.
If those affected carry out regular breathing exercises and / or respiratory therapy, it is possible to prevent an acute chest syndrome. The sick learn exercises (e.g. stretching exercises) and techniques that make breathing easier for them.
If there are any signs of acute chest syndrome, it is important to take the person to hospital immediately!
stroke
In the case of sickle cell disease, it is also possible that larger blood vessels (e.g. in the brain) constrict or clog up due to the sickle cells. For example, if a vessel in the brain is blocked, the surrounding area is no longer adequately supplied with oxygen and nutrients. A stroke occurs.
Signs of a stroke include a sudden headache, sudden numbness or weakness in one side of the face, arm or leg or even in the whole body, speech disorders or a seizure.
If the patient shows signs of a stroke, call an emergency doctor immediately!
When to the doctor
Certain symptoms indicate severe, sometimes life-threatening complications in people with sickle cell anemia. It is therefore advisable to consult a doctor immediately if:
- Sick people have a fever over 38.5 degrees Celsius
- they are pale and limp
- they have chest pain
- you are short of breath
- Joint pain occur
- they have bloating and / or abdominal pain
- the spleen is palpable and enlarged
- People suddenly get yellow eyes
- they excrete very dark urine
- they have a headache, dizziness and / or paralysis, sensory disturbances
- the penis is stiff and painful (priapism)
How do you treat sickle cell disease?
If the doctor has diagnosed sickle cell disease, it is important that the person concerned is treated by a specialized treatment team that works closely with family doctors and paediatricians. In this way, the sick person receives the best possible therapy.
The aim of treatment is to alleviate symptoms and prevent possible complaints and complications. Doctors usually prescribe medication for this (e.g. hydroxycarbamide, painkillers). In severe cases, doctors perform stem cell transplants and, in rare cases, gene therapy.
Hydroxycarbamide
Doctors usually treat patients with sickle cell disease with the active ingredient hydroxycarbamide (also: hydroxyurea). It is a cytostatic drug that is also used to treat cancer. This drug makes the blood more fluid and increases the amount of fetal hemoglobin (HbF) in the body. Fetal hemoglobin is found in healthy newborns and inhibits the formation of sickle cells.
If people with sickle cell anemia take hydroxycarbamide regularly, pain crises occur less frequently. The drug also increases the red blood pigment hemoglobin, which makes patients feel better overall.
Painkiller
People with sickle cell disease often experience severe pain, caused for example by pain crises, hand-foot syndrome and ulcers. Doctors therefore prescribe pain relievers with the active ingredients paracetamol, metamizole, diclofenac or ibuprofen. The doctors decide individually in what dosage, where (in the hospital or at home) and in what form (e.g. juice, tablet, infusion) the patients receive the medication.
People with sickle cell anemia should not be given acetylsalicylic acid (ASA). This active ingredient increases their risk of severe brain and liver damage.
Blood transfusion
Doctors prescribe blood transfusions to patients with sickle cell anemia in the event of or to prevent strokes, as well as in spleen sequestration and acute thoracic syndrome. These take place regularly (usually monthly) and often for a lifetime.
To do this, the doctors give infusions with red cell concentrates. These contain red blood cells from a healthy blood donor. This gives the affected person the urgently needed red blood cells. At the same time, the doctor replaces sickle cells with normal red blood cells in order to reduce the risk of further complications (e.g. acute chest syndrome or a stroke).
Vaccinations
To prevent life-threatening infections, doctors vaccinate young children with sickle cell anemia against pneumococci (bacteria that cause pneumonia) as early as the second month of life. Other special vaccinations that doctors recommend for sickle cell disease are vaccinations against meningococci (meningitis), haemophilus (croup, pneumonia, meningitis, joint inflammation), flu viruses and hepatitis viruses.
Antibiotics
Doctors recommend giving children with sickle cell disease from the age of three months to five months penicillin (antibiotic) every day. This helps prevent serious bacterial infections, to which people with sickle cell disease are particularly susceptible.
If patients with sickle cell anemia develop a gallbladder infection, an ulcer or an infectious disease, doctors also give antibiotics.
Stem cell transplant
A stem cell transplant (bone marrow transplant) enables doctors to cure people with sickle cell disease.
However, stem cell transplantation carries risks and is not suitable for everyone with sickle cell disease. It is therefore only carried out in patients with a very severe course of the disease.
How does the stem cell transplant work?
Doctors take stem cells from the bone marrow of a healthy donor (donor marrow) to produce healthy blood (blood stem cells). Before the transplant, doctors use chemotherapy or radiation therapy to destroy the patient's (recipient's) bone marrow, which forms the sickle cells.
The doctors then use a blood transfusion to give the sick person the healthy stem cells. In this way, they replace the diseased bone marrow with healthy stem cells and the affected person now produces new, healthy blood cells himself.
In order for the recipient's body to accept the donor's new stem cells, most of the blood must match. Doctors therefore prefer to use siblings as donors, as they often have the same characteristics (HLA genes).
Gene therapy
For some years now, doctors have been conducting studies to determine whether sickle cell disease can be permanently cured with gene therapy. To do this, they remove the affected person's stem cells and replace the pathological hemoglobin gene with a healthy gene. They then feed the genetically modified stem cells back into the person's body. This is then able to produce healthy blood cells.
So far, however, there has only been experience with individual patients in this regard. Further studies are necessary to clarify the extent to which gene therapy can be used as a treatment method for sickle cell disease.
More therapies
There are some promising drugs (e.g. preparations containing the amino acid L-glutamine) that researchers are currently testing and that may be ready for the market within the next ten years. In addition, doctors are constantly developing stem cell transplants. The so-called haploident stem cell transplant, for example, promises success, in which one parent usually serves as a donor for a sick child. Haploident means that the donor and recipient are only half the same in terms of HLA characteristics.
The treatment of people with sickle cell disease is constantly being developed. It is therefore important that sick people and their relatives inform themselves regularly about current therapies.
How does the doctor make a diagnosis?
The first point of contact if you suspect sickle cell disease is usually your pediatrician. If necessary and for further examinations, he will refer you to a specialist in internal medicine specializing in blood diseases (hematologist).
Since sickle cell anemia is a hereditary disease, the family history provides the first clues. For example, the doctor asks whether there are known carriers of the disease in the family.
Doctors also recommend that all newborns from risk regions (e.g. West and Central Africa) have a blood test carried out. Some countries do what is known as newborn screening for sickle cell anemia. A newborn screening is a general examination of all newborns in order to detect certain congenital diseases at an early stage.
Doctors also recommend checking older children and adolescents from risk areas that are anemia and often severe pain or infections for sickle cell disease. If there are indications of the disease, the doctor will do a blood test.
Blood test
To diagnose sickle cell anemia, the doctor will do a blood test. In the blood smear he can usually see the typical sickle-shaped red blood cells directly under a microscope. To do this, he takes a drop of blood from the person concerned and spreads it on a carrier (e.g. on glass). If no sickle cells can be seen, however, this does not mean that there is no sickle cell disease.
A more precise diagnosis of sickle cell disease is carried out in the laboratory using hemoglobin electrophoresis (Hb electrophoresis). It is one of the most important examination methods to detect disorders of the red blood pigment. In hemoglobin electrophoresis, the hemoglobin is dissolved from the patient's blood in a liquid and applied to a carrier. A voltage is then applied. Depending on how the hemoglobin is composed, it moves different distances in a given time. This can be used to assess whether the hemoglobin is normal or defective.
If, for example, abnormal hemoglobin (HbS) is found, which makes up less than 50 percent of the total hemoglobin in the blood, the patient is the carrier of the disease. If the person has more than 50 percent HbS in their total hemoglobin, they are likely to have sickle cell anemia.
Molecular Genetic Studies
Pathological changes (mutations) in the hemoglobin gene of the person affected can also be determined with a molecular biological examination. For example, a molecular biologist analyzes specific gene segments that are responsible for sickle cell disease from a blood or saliva sample. The polymerase chain reaction (PCR) is usually used for this. In this process, the gene segments are copied and examined with the help of an enzyme (polymerase).
Further investigations
In order to prevent symptoms of sickle cell disease or to prevent possible complications at an early stage, the following examinations are also possible:
- Examination of the child in the womb before birth (prenatal diagnosis)
- Regular check-ups in specialized centers
- Blood and urine tests
- Examination of the cerebrospinal fluid (CSF)
- Determination of the blood group if a blood transfusion is necessary
- Ultrasound of the abdomen and heart
- Ultrasound of the blood vessels in the brain (e.g. if a stroke is suspected)
- X-rays of the lungs, bones, or joints
- Computed tomography (CT) or magnetic resonance imaging (MRI)
- Examination of the eyes (e.g. if visual disturbances are suspected)