Angelman Syndrome
Lisa Vogel studied departmental journalism with a focus on medicine and biosciences at Ansbach University and deepened her journalistic knowledge in the master's degree in multimedia information and communication. This was followed by a traineeship in the editorial team. Since September 2020 she has been writing as a freelance journalist for
More posts by Lisa Vogel All content is checked by medical journalists.Angelman Syndrome (Happy Puppet Syndrome) is a rare, genetic disease. It manifests itself, among other things, through mental and physical limitations, developmental disorders (especially language) and hyperactivity. What is striking is the doll-like appearance and the happy facial expression of those affected. Find out more about the rare disease!
ICD codes for this disease: ICD codes are internationally recognized codes for medical diagnoses. They can be found, for example, in doctor's letters or on certificates of incapacity for work. Q93

Brief overview
- What is Angelman Syndrome? Rare, genetic disease that manifests itself through mental and physical limitations in child development
- Symptoms: doll-like facial features, developmental disorder, impaired coordination, no or hardly any language development, reduced intelligence, seizures, baseless laughter, laughing fits, excessive drooling, joyful waving of the arms
- Causes: genetic defect on chromosome 15
- Diagnosis: inter alia conversation, physical examination, genetic examinations
- Therapy: No causal therapy available; Supportive e.g. physiotherapy, speech therapy, occupational therapy; possibly medication to alleviate symptoms (e.g. for seizures)
- Forecast: normal life expectancy; no independent life possible
Angelman Syndrome: Definition
Angelman syndrome (AS) is caused by a genetic defect on chromosome 15. This defect disrupts the physical and mental development of those affected. Speech impairment, motor insecurity, and a happy face are the most noticeable symptoms of Angelman syndrome.
The name "Angelman Syndrome" comes from the discoverer of the disease, the English pediatrician Harry Angelman. In 1965 he compared the clinical pictures of three children who had doll-like facial features. The children laughed a lot and made jerky movements - like marionettes. This led to the English name " Happy Puppet Syndrome ”(happy puppet).
Angelman syndrome occurs in both sexes. The risk of the disease is around 1: 20,000. This makes the syndrome a rare disease.
Angelman Syndrome: Symptoms
At birth, children with Angelman syndrome are normal. Only in infancy and toddler age do the motor and cognitive developmental disorders become increasingly noticeable. Features of the genetic disorder are:
- delayed motor development
- impaired coordination
- often no or hardly any language development
- decreased intelligence
- hyperactive, high-spirited behavior
- baseless laughter
- Fits of laughter
- joyful gestures (e.g. waving your arms)
- excessive drooling
- frequent sticking out of the tongue
Some children with Angelman syndrome also have:
- Microcephaly (abnormally small head) - not at birth, but as it develops
- Seizures
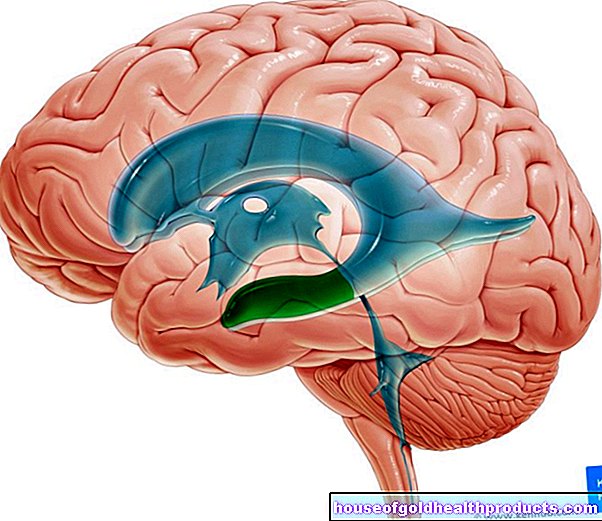
- Changes in electrical brain activity
- very light skin and eyes due to decreased pigmentation (hypopigmentation)
- Squint (strabismus)
Angelman syndrome: causes
The cause of Angelman syndrome is a genetic defect on chromosome 15: in those affected, the function of the UBE3A gene is impaired. This gene normally produces an enzyme that is involved in breaking down damaged or superfluous proteins in the cells. It thus helps the cell to work normally.
The UBE3A gene is located in chromosome region 15q11q13. There the genes are subject to what is known as “genomic imprinting”. This means that they are only active on one of the parental chromosomes (in our body cells there are two copies of each chromosome - one from the mother and one from the father). This is regulated by a chemical process - methylation: methyl groups attached at certain points can switch a gene on or off.
The gene is active on both chromosomes in numerous body cells, but not in the nerve cells of the brain: in many people there, the UBE3A gene on the paternal chromosome 15 is switched off by imprinting. As a result, UBE3A is only active on maternal chromosome 15 in the brain. This also means: If the maternal gene copy shows an error, this cannot be compensated for by the disused paternal gene copy. And it is precisely this combination that occurs in Angelman syndrome: the paternal gene segment is switched off, the maternal gene segment is defective.
The underlying genetic error can be of different types:
- Deletion: About 75 percent of all people with Angelman syndrome lack the relevant region 15q11q13 with the UBE3A gene on maternal chromosome 15. Because the corresponding region on paternal chromosome 15 is "switched off" by imprinting, the body can use the enzyme whose Construction plan is saved in Gen UBE3A, do not create.
- Mutation in the UBE3A gene: If the gene changes spontaneously, the information contained therein is lost. This is true of five to ten percent of people with Angelman syndrome. In about every fifth case there is a familial mutation: Here the mother already carries the genetic change on her father's chromosome.
- Two paternal chromosomes 15: The affected person inherited both chromosomes 15 from his father, neither from the mother (medically referred to as "paternal uniparental disomy 15"). So there is no active UBE3A gene. This applies to about one to two percent of all Angelman syndrome patients.
- Imprinting error: The UBE3A gene on maternal chromosome 15 - just like the one on paternal chromosome 15 - is switched off by imprinting. In addition, a certain part of the chromosome can be missing (deletion). An imprinting error is found in one to four percent of Angelman syndrome cases.
In the remaining ten to 15 percent of cases, the cause of Angelman syndrome is unknown. By the way: if the maternal gene is switched off and the paternal gene is defective, the children suffer from the so-called Prader-Willi syndrome.
Is Angelman Syndrome hereditary?
In general, the risk of recurrence in Angelman syndrome is low. This means the risk that parents of an affected child will have other children who also have the syndrome. In individual cases, however, this risk essentially depends on the genetic defect underlying Angelman syndrome.
For example, in Angelman syndrome due to two paternal chromosomes 15 (paternal uniparental disomy 15) it is less than one percent. On the other hand, Angelman syndrome due to an imprinting error with loss of a certain gene segment (IC deletion) can occur in half of all cases in a sibling.
This increased risk also exists with a UBE3A mutation - provided that the mother is already the carrier of the genetic defect (in around 20 percent of all mutation cases). In such cases, the mother inherited the mutation from her father. UBE3A is therefore changed in the paternal chromosome of the mother. If this is switched off, the mother will not notice the mutation. However, she can pass the chromosome on to her children, where it - then as the maternal chromosome - can cause Angelman syndrome.
Theoretically, patients with Angelman syndrome can reproduce. Depending on when the causal chromosome changes occurred (e.g. already during germ cell development or immediately after fertilization), the risk is sometimes very high (up to 100 percent) that those affected will pass the disease on. However, there is a lack of reliable data on this. There was an isolated case in September 1999: The mother who had Angelman syndrome passed the disease on here.
Angelman Syndrome: Diagnosis
If you notice the symptoms described above in your child, the pediatrician is the first point of contact. He can narrow down the possible causes more precisely and refer you and your child to a specialist if necessary.
anamnese
The first step in diagnosis is a thorough medical history. The doctor will ask you various questions about your child, such as:
- What changes did you notice in your child?
- Does your child have any physical complaints?
- Can your child sit?
- Does your child reach for objects?
- Does your child speak?
- Is your child often noticeably cheerful or excited?
- Does your child laugh in inappropriate situations, for example when they are in pain?
Physical examination
This is followed by the physical examination. The pediatrician tests the extent to which the child is regularly developing motor and mental skills. Simple exercises serve this purpose: For example, the child should concentrate on toys or specifically grab a building block. The doctor also pays attention to the child's facial expressions. Frequent laughter, doll-like features, and drooling are all signs of Angelman syndrome.
If, after the physical examination, there is a suspicion of the rare disease, the doctor will refer you to a neurologist and a human geneticist.
Genetic tests
Genetic testing is an important part of diagnosing Angelman syndrome. To do this, the doctor needs a small sample of child's cells, which he can obtain from the oral mucosa, for example, by taking a blood sample or by taking a swab. The genetic material (DNA) of these cells or the relevant chromosome region 15q11q13 is then examined in more detail in the laboratory.
In a first step, the doctors pay attention to the methylation pattern of the chromosome segment (methylation analysis / test). Further tests on the same samples (deletion analysis, mutation analysis) help to determine the cause of Angelman syndrome more closely. For this it may also be necessary to examine the genetic makeup of the parents. In this way, doctors can determine whether there is already a genetic defect there.
Further investigations
Further examinations are often useful. For example, EEG can be used to detect changes in electrical brain activity, as often occurs in Angelman syndrome. Ophthalmological examinations may also be indicated.
Angelman Syndrome: Therapy
Angelman syndrome is incurable - the genetic cause of the disease cannot be remedied. However, early intervention can have a positive effect on the motor and mental development of those affected. Regular physiotherapy, for example, is helpful. It can improve children's motor skills, alleviate restricted mobility and help prevent secondary diseases such as spinal deformities. Children with Angelman syndrome also benefit from other therapy methods such as occupational therapy and speech therapy.
In addition, some symptoms and conditions associated with Angelman syndrome may require targeted treatment. For example, anticonvulsant drugs (antiepileptic drugs) help against seizures, and tranquilizers (sedatives) for severe sleep disorders.
On the website of Verein Angelman e.V. you will find a lot of information about the Angelman syndrome, experience reports and events for those affected as well as contact persons for those affected in all regions of Germany.
Angelman syndrome: disease course and prognosis
First year of life
Babies with Angelman syndrome are more likely to have problems breastfeeding, sucking, and swallowing. They often stick their tongues out or drool a lot. In addition, children with Angelman syndrome often spit (which is often incorrectly suspected of having food intolerance or reflux disease). Frequent vomiting can lead to dangerous weight loss.
By three to six months of age, children with Angelman syndrome often start smiling. They giggle and gurgle a lot and often stick their tongues out during these outbursts of joy.
The delayed motor development is usually noticeable between the ages of 6 and 12 months: the children do not crawl or sit. The movements of the upper body are often shaky. This in turn makes sitting more difficult.
A fraction of those affected already have seizures at the age of 12 months.
One to three years
Within the first three years of life, the developmental disorder in Angelman syndrome becomes very evident. The children suffer more from mild seizures. Some of them are hyperactive, overexcited and always on the move. Many tend to put their hands or toys in their mouths all the time, or stick their tongues out frequently and drool. If the children are particularly excited, they often laugh excessively and wave with outstretched arms.
The impaired language development becomes apparent for the first time at this age. Children “babble” to themselves or scream and squeak, however, can only pronounce a few or no easily understandable words and usually use them without context.But they understand language and there is also social interaction with other people.
Puberty and adulthood
Puberty is often three to five years late in children with Angelman syndrome. However, sexual maturity then develops normally. There is still no language development if language comprehension is often present. The seizures in adulthood can usually be managed well with medication.
People with Angelman syndrome have a normal life expectancy. An independent life is not possible due to the mental limitations.
Tags: drugs teeth news













.jpg)