Spinal muscular atrophy
and Florian Tiefenböck, doctor Updated onMaximilian Reindl studied chemistry and biochemistry at the LMU in Munich and has been a member of the editorial team since December 2020. He will familiarize himself with medical, scientific and health policy topics for you in order to make them understandable and comprehensible.
More posts by Maximilian ReindlFlorian Tiefenböck studied human medicine at the LMU Munich. In March 2014, he joined as a student and has supported the editorial team with medical articles ever since. After receiving his medical license and practical work in internal medicine at the University Hospital Augsburg, he has been a permanent member of the team since December 2019 and, among other things, ensures the medical quality of the tools.
More posts by Florian Tiefenböck All content is checked by medical journalists.
Spinal muscular atrophy, or SMA for short, is a rare disease in which certain nerve cells in the spinal cord die. Stimuli and impulses from the brain then no longer reach their destination: the muscles. This causes muscle wasting and paralysis. There are different forms of SMA. The most severe begins in infancy. New treatments promise a lasting improvement in health. Read more about spinal muscular atrophies here.
ICD codes for this disease: ICD codes are internationally recognized codes for medical diagnoses. They can be found, for example, in doctor's letters or on certificates of incapacity for work. G12

Brief overview
- What is spinal muscular atrophy? A group of muscle weakness diseases. They are based on the death of certain nerve cells in the spinal cord that control the muscles (motor neurons). Therefore, the SMA are among the motor neuron diseases.
- What forms are there? Hereditary spinal muscular atrophies are mostly SMA with a certain genetic defect on chromosome 5 (5q-associated SMA). Doctors distinguish between four different forms: SMA type 1 – type 4. In addition, there are sporadic forms, the heredity of which is not certain.
- Frequency: Rare Disease; the inherited SMA affect about one newborn in 7000.
- Symptoms: muscle twitching, progressive muscle weakness, muscle wasting, paralysis. Gradients differ depending on the SMA shape.
- Causes: The hereditary spinal muscular atrophy types 1-4 are the result of a genetic defect on chromosome 5, more precisely on the SMN1 gene. As a result, the body lacks a special protein, the SMN protein. This deficit damages the motor neurons in the spinal cord.
- Diagnosis: Genetic examination for altered SMN genetic makeup, physical examinations, electroneurography, electromyography, blood tests (e.g. CK)
- Treatment: Gene replacement therapy or drug administration of splicing modulators are possible. Accompanying physiotherapy, speech therapy, pain therapy and psychotherapy. If necessary, surgery on the spine. Treatment plan depending on the type of SMA.
- Prognosis: In the case of hereditary proximal SMA, new therapy options have a causal effect and can have a positive effect on the course of the disease. Starting treatment early is crucial. Treatments are not yet available to every patient. If left untreated, children with SMA type 1 usually die within the first two years. Life expectancy in type 3 and type 4 hardly or not reduced.
What is spinal muscular atrophy?
In spinal muscular atrophy (SMA), certain nerve cells in the spinal cord die. They usually control muscles, which is why experts refer to these nerve cells as motor neurons. Accordingly, the SMA belong to the so-called motor neuron diseases.
In spinal muscular atrophies, the lower (second) motor neurons, which are directly connected to the muscles with their appendages, are affected. As a result of the damage, fewer or no more nerve signals reach the muscles. The muscles become increasingly weaker and smaller (muscle wasting / muscle atrophy).
Doctors differentiate between different forms of spinal muscular atrophy. By far the largest group are the hereditary SMA, in which the muscles close to the trunk (proximal) are affected. They are based on a specific genetic defect. About one in 7,000 newborns will develop it.
Spinal muscular atrophy is a rare disease overall. Nonetheless, it is the second most common autosomal recessive inherited disease. It is also the most common cause of death in babies or toddlers due to a genetic defect.
What types of spinal muscular atrophy are there?
Doctors differentiate hereditary (hereditary) forms of SMA from sporadic forms. Another classification of spinal muscular atrophy relates primarily to the muscle groups affected first. There are
- Proximal SMA: With around 90 percent, they form the largest SMA group. The symptoms begin in the muscles near the trunk, i.e. proximally.
- Non-proximal SMA: Here, more distant muscle groups, such as the hands and feet, are affected first (distal SMA). In the further course, these SMA can also spread to muscles near the middle of the body.
- Special forms (e.g. Kennedy type spinobulbar muscular atrophy)
Proximal spinal muscular atrophies
Hereditary proximal spinal muscular atrophies are mostly diseases that are based on a specific genetic defect (5q-associated SMA, defect on chromosome 5). These in turn are divided into four different forms. The classification is based on the time at which the first symptoms appear and on the course of the disease.
Spinal Muscular Atrophy Type 1: This is the most common and most serious form of SMA. It is also called "Werdnig-Hoffmann's disease" or "acute infantile SMA". The disease usually begins in early infancy. The muscle weakness affects the whole body - doctors also speak of a "floppy infant syndrome" (from English floppy = flaccid, infant = infant, child). Most untreated children with SMA type 1 die before they are two years old.
Spinal muscular atrophy type 2: This form of SMA is also known as "intermediate spinal muscular atrophy" or "chronic infantile SMA". The first symptoms typically appear before the age of 18 months. Those affected have a sometimes significantly reduced life expectancy.
Spinal muscular atrophy type 3: It is also known as "juvenile spinal muscular atrophy" or "Kugelberg-Welander's disease". This SMA usually begins after the age of 18 months and before early adulthood. The muscle weakness is milder than in type 1 or 2. Those affected have only a slightly reduced life expectancy.
Spinal muscular atrophy type 4: It is similar to SMA type 3, but only appears in adulthood (usually> 30 years). However, the muscle weakness is less pronounced and progresses more slowly than with SMA type 3.
The transitions between the different versions are fluid. In some cases, this makes a clear delimitation difficult. Some genetic predispositions also play an important role in the severity of the disease in question.
Other spinal muscular atrophies
In addition to these proximal forms, there are other forms of spinal muscular atrophy. These include, for example, rarer, also hereditary distal spinal muscular atrophies. With them, the symptoms typically begin in muscle groups that are further away from the body.
Inheritance is not guaranteed in the case of sporadic SMA. In addition, no familial accumulation can be determined. In the literature, these include:
- Hirayama type (juvenile distal SMA, disease around the age of 15, affects arm muscles, usually comes to a standstill even without therapy and can even improve)
- Vulpian-Bernhard type (also known as "flail-arm" syndrome with onset in the shoulder girdle, usually from the age of 40)
- Duchenne-Aran type (initially affected hand muscles, spreading towards the trunk of the body, usually after the age of 30)
- Peroneal type ("flail-leg" syndrome, first on the lower leg muscles)
- Progressive bulbar palsy (speech and swallowing disorders, affects about 20 percent of patients with amyotrophic lateral sclerosis)
Some sporadic SMA forms ("flail-arm - / - leg" syndrome, progressive bulbar paralysis) are counted among the variants of amyotrophic lateral sclerosis (ALS) in specialist circles. This article deals primarily with hereditary proximal spinal muscular atrophies.
Spinobulbar muscular atrophy
Spinobulbar or bulbospinal muscular atrophy (Kennedy type, Kennedy syndrome) is an inherited disease. It often begins in young to middle adulthood. This special SMA form is inherited in an X-linked recessive manner and therefore only affects men (since men only have one X chromosome, in women the second, healthy X chromosome predominates and would compensate for the defect).
Muscle weakness in the muscles close to the body on the legs and arms or shoulders as well as the tongue and throat muscles is common. As a result, those affected have problems speaking and swallowing, for example. They also complain of tremors, muscle cramps and twitching. Affected men also often have stunted testicles and are sterile. In addition, the mammary glands enlarge (gynecomastia).
Spinobulbar muscular atrophy is usually slow. Life expectancy is hardly restricted.
How do you recognize spinal muscular atrophy?
Typical of spinal muscular atrophy are progressive muscle weakness up to paralysis (paresis) and muscle twitching. As a result of the nerve damage, the muscles no longer receive electrical impulses, as a result of which they shrink over time (muscle atrophy). The exact signs and complaints depend on the respective form. The following section looks at the symptoms of hereditary proximal SMA.
Symptoms of Infantile Spinal Muscular Atrophy Type 1
With SMA type 1, symptoms appear in the first six months of life. Generalized muscle weakness occurs - that is, one that affects the whole body. In addition, the tension between the muscles decreases. Doctors speak of hypotonia.
In newborns, this muscle weakness initially manifests itself in a typical leg posture, which is reminiscent of a lying frog (frog leg posture). The legs are bent, the knees angled outwards and the feet angled inwards. In most cases, it is also not possible to lift or hold the head on your own.
At an advanced age, children with SMA type 1 cannot sit or walk on their own. Many children are also unable to speak, as the tongue muscles can also be affected.
Another characteristic of type 1 spinal muscular atrophy is the shape of the upper body: the muscles of the chest and back do not develop properly. This gives the upper body a bell-like shape (bell chest). Due to the low development of the muscles in the chest and back, those affected adopt a hunched posture.
Often there is also an increasing curvature of the spine (scoliosis). The hunched forward and hunched posture causes further breathing problems. Very fast and shallow breathing (tachypnea) is characteristic.
Symptoms of intermediate spinal muscular atrophy type 2
Type 2 spinal muscular atrophy usually only causes symptoms between the ages of seven and 18 months. Affected children can sit on their own, but usually neither learn to stand nor to walk. The muscle weakness progresses more slowly than in type 1.
With SMA type 2, symptoms similar to those of the severe infantile form, such as deformation of the spine, arise over time. The joints stiffen due to shortened muscles and tendons (contractures). Other signs include tremors in the hands and twitching of the muscles in the tongue.
Symptoms of Juvenile Spinal Muscular Atrophy Type 3
Type 3 spinal muscular atrophy typically appears after the age of 18 months and before the age of 18. Affected children can sit, stand and walk independently. However, the muscle weakness, especially in the pelvic and leg muscles, causes a waddling gait.
Over the course of several years, performance decreases: At first, those affected find sporting activities or climbing stairs difficult, but finally it is also difficult to carry shopping bags, for example. After many years, type 3 spinal muscular atrophy makes running and any other exertion difficult or impossible, even in older people.
Overall, however, the symptoms are less pronounced than in the two other forms of disease, type 1 and type 2. For many of those affected, the quality of life is hardly impaired over a long period of time.
Symptoms of adult spinal muscular atrophy type 4
This very rare form of progressive muscle wasting begins in adulthood, often from the third decade of life. Initially, the leg and hip muscles are affected. As the disease progresses, muscle weakness also spreads to the shoulders and arms.
The manifestation of the clinical picture is similar to that of juvenile SMA type 3. However, the progressive muscle weakness is even slower than with SMA type 3.
What causes spinal muscular atrophy?
In spinal muscular atrophy, the second motor neurons in the spinal cord perish. These are nerve cells that control the muscles with their appendages. As a result of the damage to these highly specialized motor neurons, fewer electrical signals reach the muscles than is the case in healthy people. If muscle cells are used less and therefore less stimulated, the body breaks them down over time.
Genetic defect
In most cases, spinal muscular atrophy is a hereditary disease (hereditary SMA). The cause of the typical proximal SMA forms is incorrect information in the patient's genetic make-up. The so-called SMN1 gene on chromosome 5 is not functional.
The SMN1 gene carries the information - i.e. the blueprint - for the vital protein molecule called SMN. SMN stands for "Survival (of) Motor Neuron". Without the SMN protein molecule, the motor neurons perish over time.
It is true that there is also a related SMN2 gene in the body, which in principle is able to “compensate” for the non-functional SMN1 genetic information. But this usually only happens to a small extent. This means that a loss of function of the SMN1 gene (if left untreated) can usually not be completely compensated for by an intact SMN2 gene copy.
Autosomal recessive and autosomal dominant inheritance
A person's genetic information is available in duplicate. As a result, everyone has two copies of the SMN1 gene - one from their father and one from their mother. The proximal spinal muscular atrophies of childhood are typically inherited as an autosomal recessive trait.
This means that both gene variants (alleles) from the parents must be defective for spinal muscular atrophy to develop in the offspring. In the case of recessive inheritance, the parents are not affected because, in addition to the functionless one, they also have a healthy SMN1 gene that compensates for the defect.
Around every 45th person is the owner of this system for SMA. A couple in which both partners are carriers has a 25% risk of having a child with the disease.
In a few cases in adolescence, spinal muscular atrophies in adulthood in particular also follow an autosomal dominant inheritance. In the case of dominant inheritance, a defective gene already asserts itself - and those affected become ill. However, this is not the aforementioned genetic defect on chromosome 5. These 5q-associated SMAs are always inherited in an autosomal recessive manner.
Inheritance with other SMA forms
Non-proximal spinal muscular atrophy can also be inherited. The special spinobulbar form (Kennedy type) is inherited recessively via a sex chromosome, the X chromosome (this affects the gene variants that contain the blueprint for docking sites for male sex hormones). In the case of sporadic forms, however, inheritance is not guaranteed. Little is known here why exactly the second motor neurons perish.
Examinations and diagnosis
The diagnosis of spinal muscular atrophy is usually made by paediatricians, pediatricians who specialize in nerve diseases (neuropediatricians) and specialists in diseases of the nervous system (neurologists). Various examinations are necessary for a more precise clarification. In the case of an SMA, genetic tests and examinations of the nerves and muscles are particularly important.
Collection of the medical history (anamnesis)
With every illness, the doctor first asks about the symptoms that have occurred and how it has progressed so far. In babies and toddlers, parents report changes and abnormalities in their child's behavior. In the case of hereditary diseases in particular, doctors also focus on the family's medical history.
Physical examinations
Basically, a doctor determines abnormalities in motor development by physically examining the child. For example, it tests whether the children can independently hold their heads upright, sit or move their arms or legs independently (depending on their age).
Complementary exercise tests are carried out in older children and adults with suspected spinal muscular atrophy. The doctor checks how much strength the person concerned can muster and for how long he can hold it. He also examines endurance.
In addition, the doctor tests the reflexes, which are typically weakened or extinguished, especially in the case of pronounced spinal muscular atrophies. To do this, he taps various tendons with a hammer, for example on the heel or under the knee, and checks the reaction.
Genetic Studies
The most reliable detection method for (hereditary) spinal muscular atrophy is genetic analysis. Doctors look for evidence of an altered (mutated) SMN1 gene and the number of existing SMN2 copies.
The general rule is to diagnose and treat (hereditary) SMA as early as possible. Depending on the form and available treatment, motor development can be positively influenced before the motor neurons of the spinal cord are irreversibly damaged.
Further investigations at SMA
If an SMA is suspected, doctors often measure the conduction speed of the nerves (electroneurography) and muscle activity (electromyography). If necessary, they also examine the muscles using ultrasound (myosonography) or magnetic resonance imaging (MRI).
In addition, doctors arrange blood tests. If there is spinal muscular atrophy, certain parameters can be changed: for example, the level of creatine kinase (CK, a typical muscle enzyme) is increased.
Treatment of spinal muscular atrophy
Treatment for spinal muscular atrophy is complex. For a long time, causal therapy was not possible for any form of SMA. However, advances in medical research are giving doctors new treatment options to fundamentally help those affected with proximal SMA (SMN gene defect on chromosome 5).
In addition, doctors concentrate on alleviating the symptoms and providing the best possible support to those affected (e.g. physiotherapy, respiratory therapy, psychotherapy, possibly surgery).
Medical therapy
The new treatment approaches for patients in whom the SMA is based on a known SMN gene defect intervene directly in the genetic material itself or in the downstream processing of the genetic information.
The aim is to enable the patient's body to independently produce sufficient quantities of the SMN protein, which is crucial for the motor neurons.
The following treatment options are available for spinal muscular atrophy:
- Splicing modulators (Nusinersen, Risdiplam): These drugs intervene directly in the further processing of messenger RNA molecules. They strengthen those processes that deliver a higher amount of SMN protein from the intact SMN2 gene.
- Gene replacement therapy (Onasemnogene Abeparvovec): This therapy intervenes directly in the human genome. The faulty copy of the SMN1 gene is replaced in the affected cells by an externally supplied, functional gene construct.
Splicing modulators
In the case of an SMN1 gene defect, the body can alternatively produce the SMN protein from the related SMN2 gene. The replacement gene SMN2 “jumps in”, but that is not enough. The reason: The proteins in SMN2 are usually too short and are broken down quickly.
This is due to the processing of the corresponding SMN2 messenger RNA (SMN2 mRNA). It transmits the construction information from the genome (DNA) to the protein production sites (ribosomes).
To do this, the SMN2 gene in the genome is first read. A preliminary SMN2 messenger RNA is produced. Among other things, it has to be processed further by so-called splicing. Only then does the mature messenger RNA arise. Special cell complexes, the ribosomes, then read the mature messenger RNA and thus produce SMN2 protein. And it is precisely this that is shortened and unstable, is quickly dismantled and cannot take over the function of SMN1.
To change this, the active ingredients Nusinersen and Risdiplam influence the further processing of the preliminary messenger RNA. As a result, these so-called splicing modulators ultimately increase the amount of usable SMN proteins - and can thus ensure an adequate supply.
Nusinersen
The drug Nusinersen is a so-called "antisense oligonucleotide" (ASO). It was approved by the European Medicines Agency in 2017. ASO are artificially produced and specially adapted RNA molecules. They bind specifically and precisely to the SMN2 messenger RNA. This prevents them from being further processed incorrectly in the human cell.
Specifically: Nusinersen prevents important information (exon 7) from being erroneously cut out of the SMN2 messenger RNA. The whereabouts of exon 7 causes the body to subsequently make more functional SMN protein.
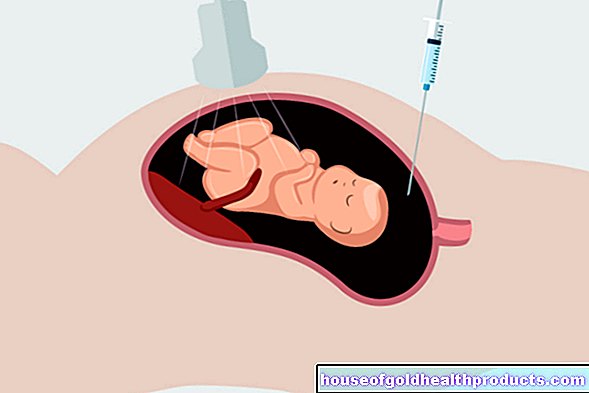
Nusinersen is given through what is known as a lumbar puncture. This means that the drug is injected into the spinal canal with a syringe. This therapy is repeated at regular intervals of a few months. In the first year of treatment, those affected receive six, then three doses a year.
Patients usually tolerate the drug well. Nusinersen leads to more favorable disease courses. Studies have shown that mobility improves in many patients: in many cases, it was possible to sit freely and turn the body independently. Side effects and complications are based, among other things, on the lumbar puncture (e.g. headache, infections of the meninges).
Risdiplam
In March 2021, the European Commission approved Risdiplam as the third drug against 5q-associated SMA (types 1-3 or one to four SMN2 gene copies). Risdiplam is taken daily as a dissolved powder. The exact dose is calculated based on age and body weight.
Unlike Nusinersen, Risdiplam is not an "antisense oligonucleotide", but a small molecule. This molecule binds to the messenger RNA for SMN2 proteins and stabilizes them in this way. As a result, more functional SMN proteins are created.
Common side effects of Risdiplam include gastrointestinal discomfort, rash, fever, and urinary tract infections.
Gene replacement therapy
Another approach to the treatment of proximal spinal muscular atrophy relies on what is known as gene replacement therapy. The defective SMN1 gene - the starting point of the progressive SMA - is "replaced" by a new functional copy of the gene.
The active ingredient Onasemnogene Abeparvovec (AVXS-101), which works on this principle, received conditional marketing authorization for the treatment of toddlers and children from the European Medicines Agency (EMA) in May 2020.
The drug can be used for SMA type 1 according to EMA information. In all other forms of SMA disease, genetic characteristics (number of SMN2 copies) decide whether gene replacement therapy is an option.
With Onasemnogene Abeparvovec, a functional copy of the human SMN1 gene is introduced into the affected cells of the spinal cord and the brain stem. This is done by certain viruses that serve as “ferries” for the new genetic material - so-called adeno-associated viral vectors (AAV vectors).
The vector gene constructs are given once as an infusion via the vein into the bloodstream and from there are distributed throughout the body. Due to a not yet fully developed blood-brain barrier in small children, these vectors can also get into the spinal cord tissue.
By preferentially binding these vectors to special surface structures of the motor neurons, they preferentially take up the genetic material in order to then produce the SMN protein independently.
Treatment can improve motor functions and lead to lasting developmental success (e.g. sitting, crawling and walking without support).During the treatment, the liver values can sometimes increase significantly, but the number of blood platelets can decrease. Fever and vomiting are also common. To reduce side effects, patients are given corticosteroids ("cortisone") for a few weeks.
Age-appropriate motor development is generally only possible if gene therapy has started presymptomatically. Treatment takes place in specialized neuromuscular treatment centers.
physical therapy
Physiotherapy continues to be an important pillar of the treatment of SMA. Not every form of SMA can be treated with the new treatment approaches. Regular exercise therapy is designed to maintain physical abilities and slow down muscle breakdown.
The physiotherapist passively moves parts of the body that are already paralyzed. Active movement sequences, in turn, are trained to support the mobility and strength of the muscles. Massage or heat and cold treatments can also help. These also serve to relax and, under certain circumstances, slow down further degeneration.
Speech therapy
In some cases, SMA affects the speaking and swallowing muscles. Then a speech therapy exercise helps. It encourages children to learn to speak. Even in older patients, this can usually slow down a deterioration in speech. Speech therapists also train correct swallowing.
Both physiotherapists and speech therapists support those affected with targeted breathing therapy.
Pain Relieving Treatment
Pain therapy plays an important role, especially in the more advanced stages of the disease. Doctors use pain reliever medication to reduce the suffering of those affected.
surgery
Since spinal muscular atrophy can lead to severe curvature of the spine (scoliosis), doctors sometimes consider surgery. In doing so, they specifically stiffen the spine.
This gives those affected a (certain) additional trunk stability, which not only enables a more upright posture, but also protects bones and joints. A spinal surgery can also help against progressive breathing problems.
Psychotherapeutic care
Neuromuscular diseases such as spinal muscular atrophy represent great psychological stress. Patients and relatives process the diagnosis in individual and group sessions led by psychotherapy and develop strategies to better deal with the disease.
Self-help groups and patient representatives also offer important support. They inform, advise and support those affected and their relatives to cope with the challenges of an SMA disease.
Chances of recovery from spinal muscular atrophies
If there is spinal muscular atrophy, the prognosis depends primarily on the respective shape. The later the symptoms appear, the better the course. In addition, the earlier doctors diagnose spinal muscular atrophy, the sooner they can initiate suitable treatment measures, even before motor neurons have been irreversibly damaged.
The new treatment options through splicing modulators and gene replacement therapy hold great potential in the treatment of proximal SMA - especially if treatment is started (very) early. However, data for a reliable long-term prognosis are still pending. Only further studies and close-knit drug safety observations can provide further certainty over the next (months and) years. With the newer drugs, long-term control of the disease or even a cure are at least conceivable.
SMA type 1 is generally a serious disease. Children who develop SMA type 1 have (untreated) a very limited life expectancy. The rapidly increasing muscle weakness all over the body also affects breathing. The consequences are acute pneumonia and even respiratory failure. Affected children die within the first few years of life.
The prognosis is slightly better for SMA type 2. Life expectancy varies depending on the exact severity of the disease: some die in childhood, but most of them reach young adulthood. Sooner or later - if desired - breathing must be supported in the more severe forms. Affected people stay mobile with the help of a wheelchair.
With SMA type 3, the prognosis is significantly better - especially if the first symptoms appear late. The performance gradually deteriorates over the course of several years. In old age, a wheelchair or even permanent care may be necessary. Life expectancy is hardly restricted by type 3 spinal muscular atrophy.
Adult spinal muscular atrophy (type 4) is even slower than type 3. People usually have a normal life expectancy.
Tags: sex partnership prevention drugs





















-kopfsache.jpg)